
Modulation of Inflammatory Cytokine Production in Human Monocytes by cGMP and IRAK3


 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
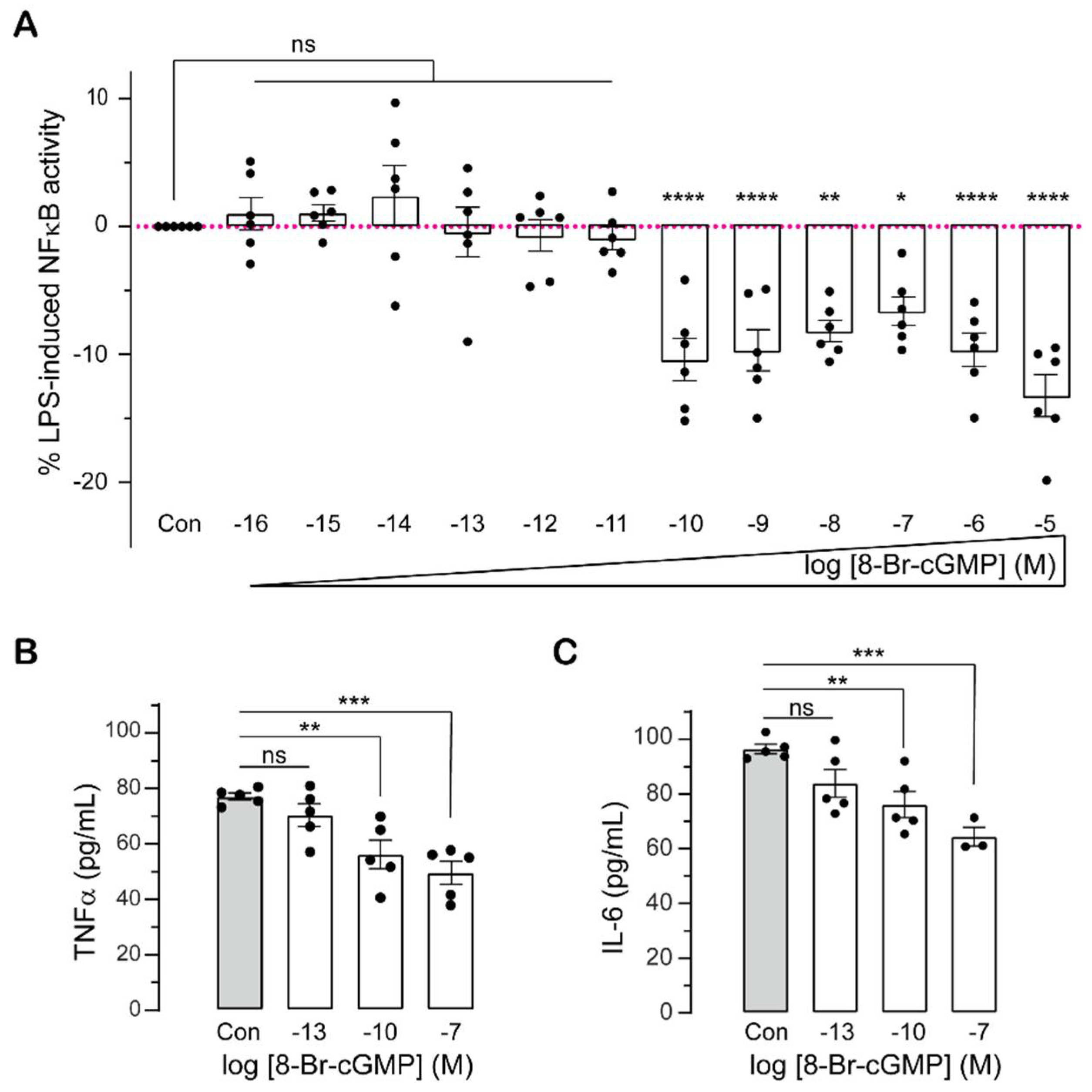
2.1. Sub-Nanomolar cGMP Suppresses Cytokine Production in LPS-Challenged Monocytes
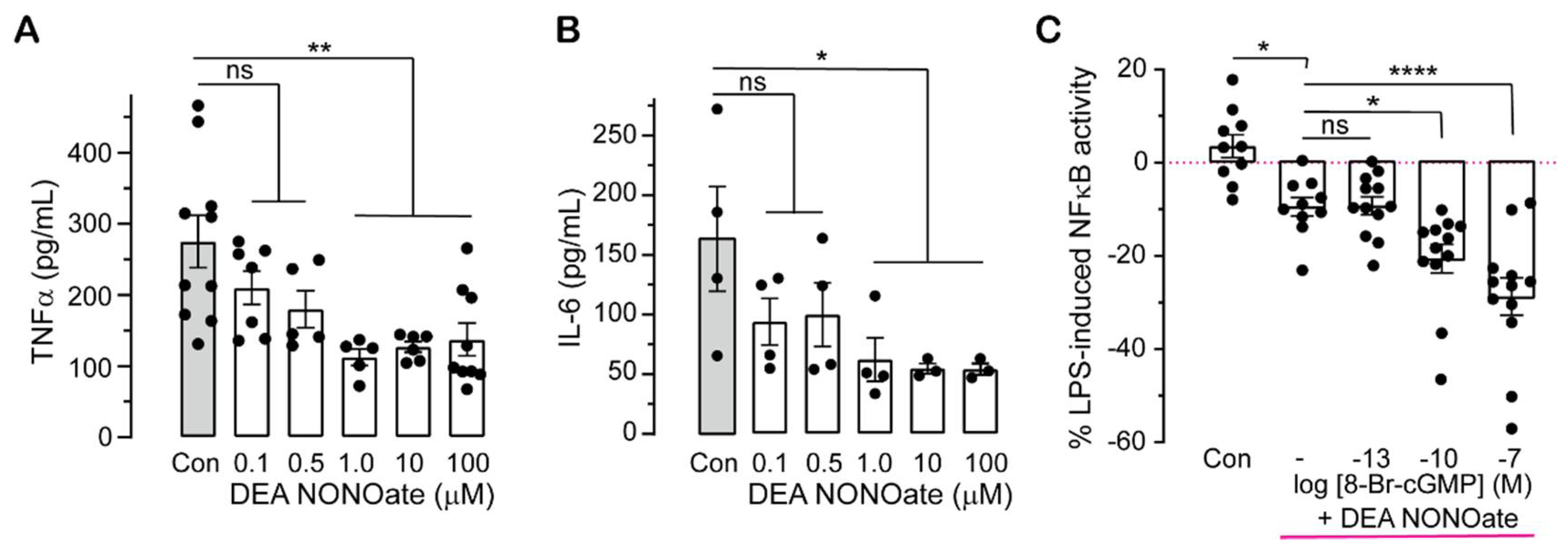
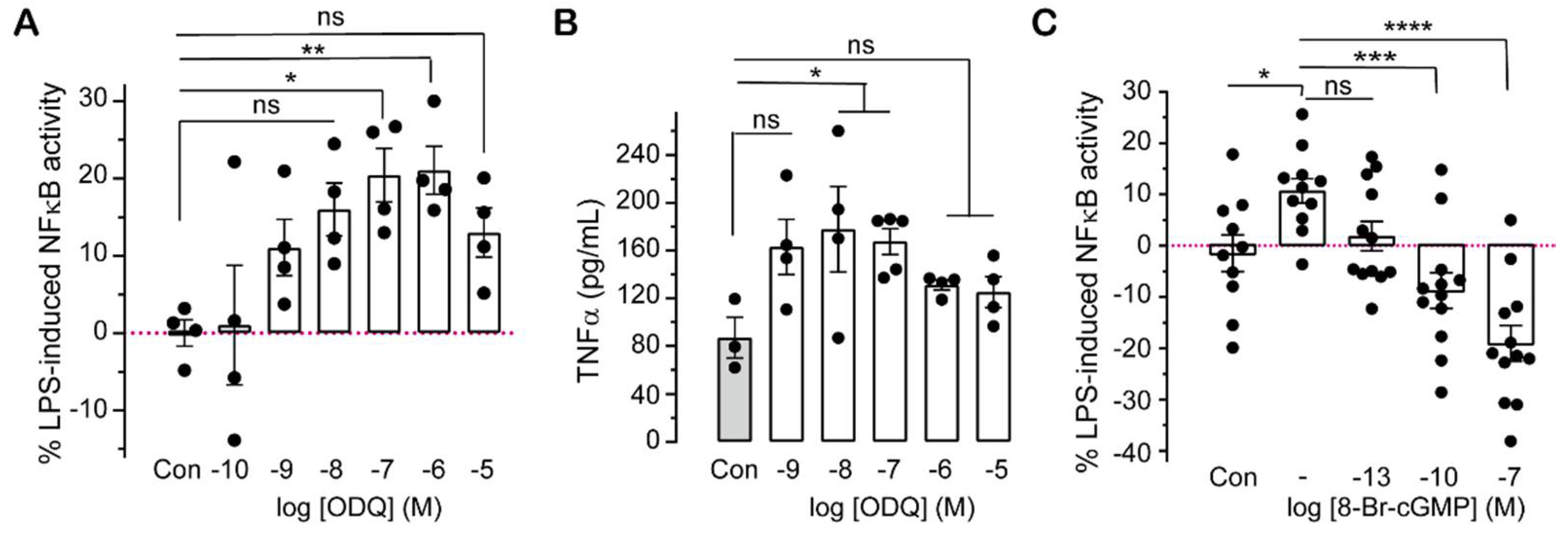
2.2. Pharmacologically Altering cGMP Levels Changes Inflammatory Responses
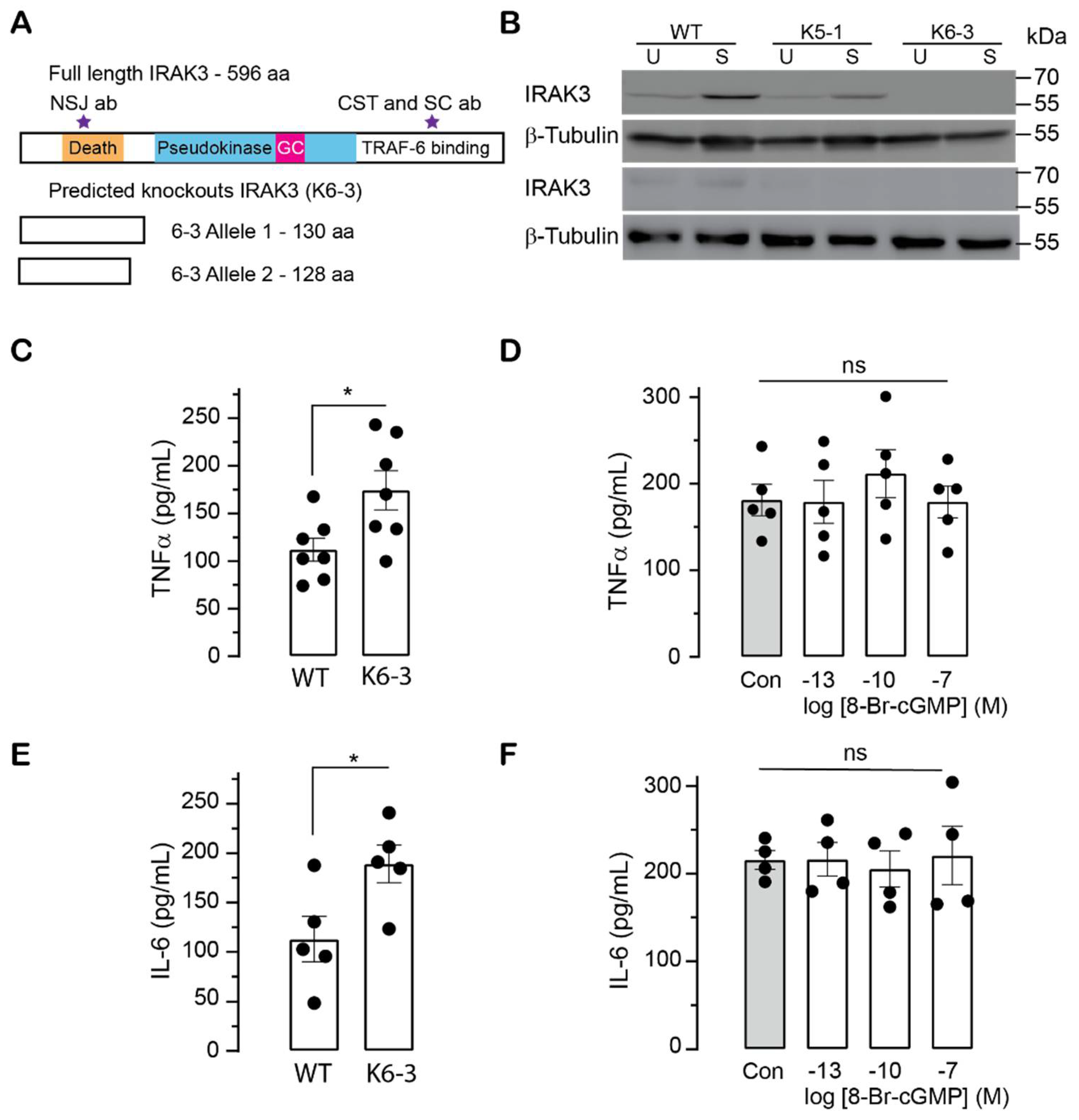
2.3. Effects of cGMP on Inflammatory Markers in Wild Type and IRAK3 Knockdown/Knockout THP-1 Cells
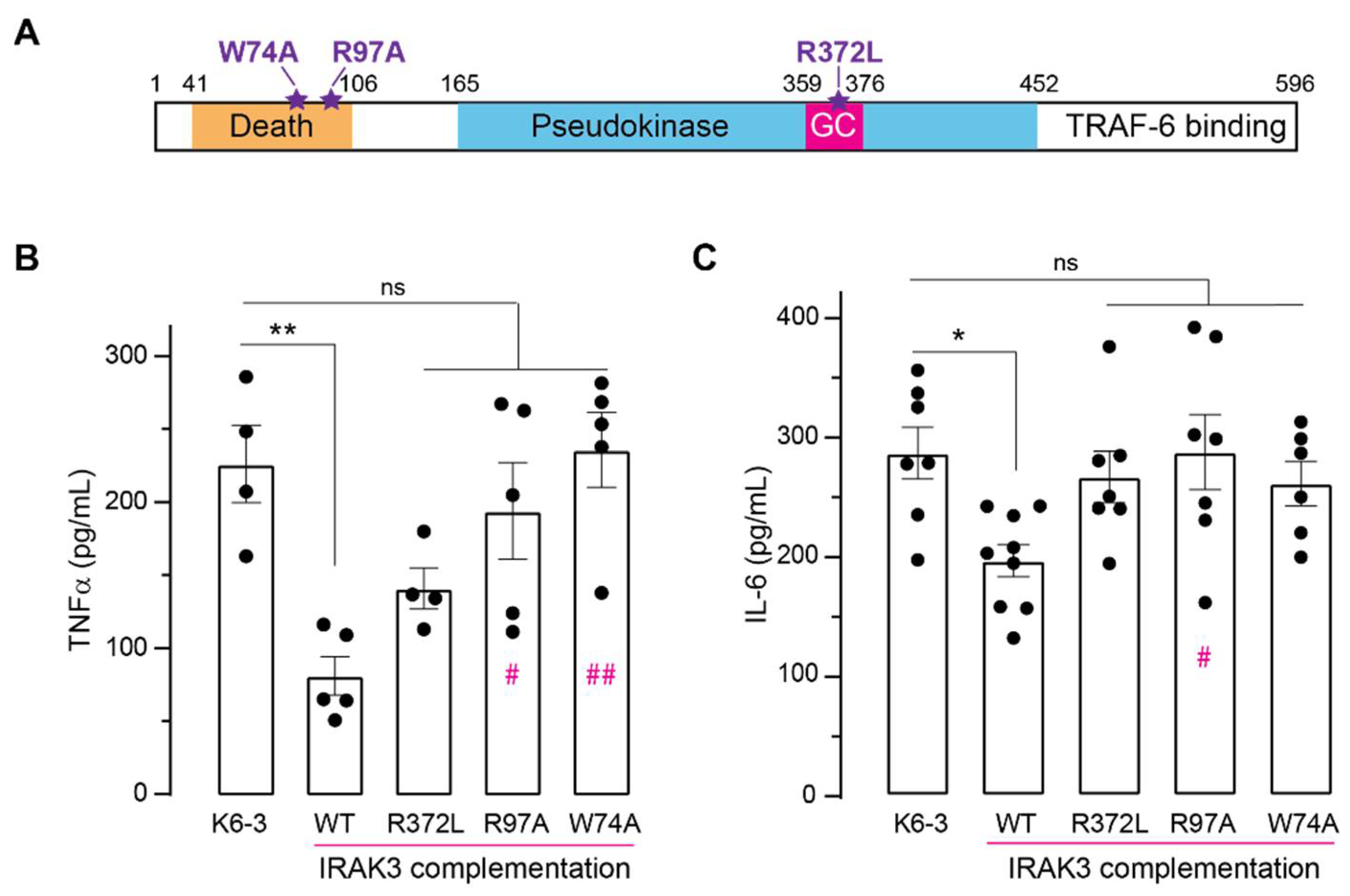
2.4. Effect of Mutations in IRAK3 on Cytokine Level
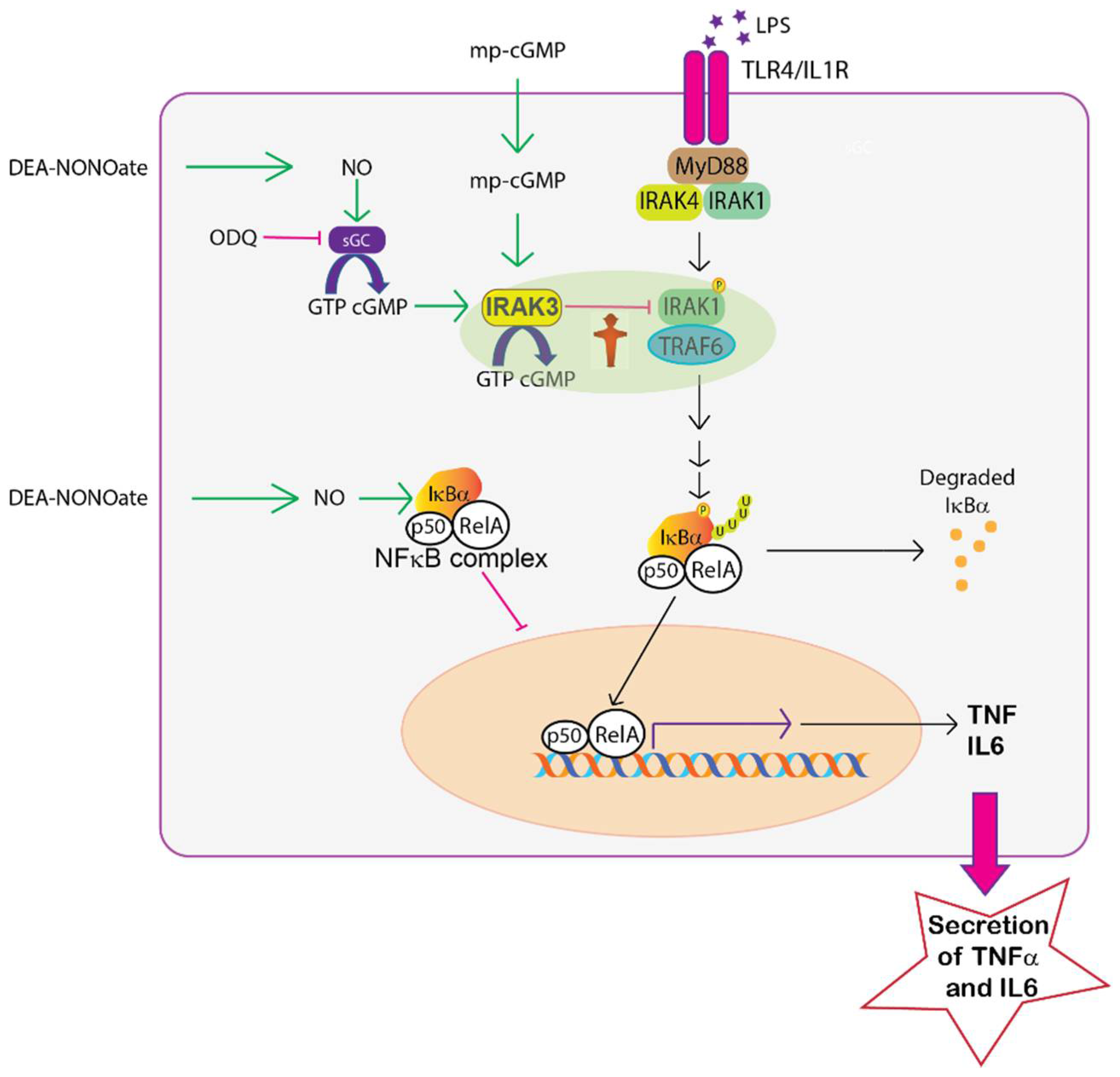
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. SEAP Assay
4.3. ELISA
4.4. Immunoblots
4.5. Generating IRAK3 Gene Knockdown THP-1 Cells
4.6. Transfection of Wild Type and Mutant IRAK3 Constructs to IRAK3 Knockdown Cells
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nathan, C. Points of control in inflammation. Nature 2002, 420, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Monneret, G.; Payen, D. Sepsis-induced immunosuppression: From cellular dysfunctions to immunotherapy. Nat. Rev. Immunol. 2013, 13, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Hernandez, L.D.; Galan, J.E.; Janeway, C.A.; Medzhitov, R.; Flavell, R.A. IRAK-M is a negative regulator of toll-like receptor signaling. Cell 2002, 110, 191–202. [Google Scholar] [CrossRef] [Green Version]
- Miyata, M.; Lee, J.-Y.; Susuki-Miyata, S.; Wang, W.Y.; Xu, H.; Kai, H.; Kobayashi, K.S.; Flavell, R.A.; Li, J.-D. Glucocorticoids suppress inflammation via the upregulation of negative regulator IRAK-M. Nat. Commun. 2015, 6, 6062. [Google Scholar] [CrossRef] [Green Version]
- Wesche, H.; Gao, X.; Li, X.; Kirschning, C.J.; Stark, G.R.; Cao, Z. IRAK-M is a novel member of the pelle/interleukin-1 receptor-associated kinase (IRAK) family. J. Biol. Chem. 1999, 274, 19403–19410. [Google Scholar] [CrossRef] [Green Version]
- Rhyasen, G.W.; Starczynowski, D.T. IRAK signalling in cancer. Br. J. Cancer 2015, 112, 232–237. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Nicolaes, G.A.; Kruijswijk, D.; Versloot, M.; van der Poll, T.; van’t Veer, C. The structure function of the death domain of human IRAK-M. Cell Commun. Signal. 2014, 12, 77. [Google Scholar] [CrossRef]
- Jain, A.; Kaczanowska, S.; Davila, E. IL-1 receptor-associated kinase signaling and its role in inflammation, cancer progression, and therapy resistance. Front. Immunol. 2014, 5, 553. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.C.; Lo, Y.C.; Wu, H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature 2010, 465, 885–890. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Yu, M.; Fukuda, K.; Im, J.; Yao, P.; Cui, W.; Bulek, K.; Zepp, J.; Wan, Y.; Kim, T.W.; et al. IRAK-M mediates Toll-like receptor/IL-1R-induced NF-κB activation and cytokine production. EMBO J. 2013, 32, 583–596. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.H.; Turek, I.; Meehan-Andrews, T.; Zacharias, A.; Irving, H. Analysis of interleukin-1 receptor associated kinase-3 (IRAK3) function in modulating expression of inflammatory markers in cell culture models: A systematic review and meta-analysis. PLoS ONE 2020, 15, e0244570. [Google Scholar] [CrossRef] [PubMed]
- Nechama, M.; Kwon, J.; Wei, S.; Kyi, A.T.; Welner, R.S.; Ben-Dov, I.Z.; Arredouani, M.S.; Asara, J.M.; Chen, C.H.; Tsai, C.Y.; et al. The IL-33-PIN1-IRAK-M axis is critical for type 2 immunity in IL-33-induced allergic airway inflammation. Nat. Commun. 2018, 9, 1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Yu, M.; Zhao, J.; Martin, B.N.; Roychowdhury, S.; McMullen, M.R.; Wang, E.; Fox, P.L.; Yamasaki, S.; Nagy, L.E.; et al. IRAK-M-mincle axis links cell death to inflammation: Pathophysiological implications for chronic alcoholic liver disease. Hepatology 2016, 64, 1978–1993. [Google Scholar] [CrossRef] [Green Version]
- Freihat, L.; Wheeler, J.; Wong, A.; Turek, I.; Manallack, D.; Irving, H. IRAK3 modulates downstream innate immune signalling through its guanylate cyclase activity. Sci. Rep. 2019, 9, 15468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freihat, L.; Muleya, V.; Manallack, D.T.; Wheeler, J.I.; Irving, H.R. Comparison of moonlighting guanylate cyclases: Roles in signal direction? Biochem. Soc. Trans. 2014, 42, 1773–1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, M.; Loughran, P.A.; Zhang, L.; Scott, M.J.; Billiar, T.R. Shedding of the tumor necrosis factor (TNF) receptor from the surface of hepatocytes during sepsis limits inflammation through cGMP signaling. Sci. Signal. 2015, 8, ra11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beavo, J.A.; Brunton, L.L. Cyclic nucleotide research—Still expanding after half a century. Nat. Rev. Mol. Cell Biol. 2002, 3, 710–717. [Google Scholar] [CrossRef]
- Kniotek, M.; Boguska, A. Sildenafil can affect innate and adaptive immune system in both experimental animals and patients. J. Immunol. Res. 2017, 2017, 4541958. [Google Scholar] [CrossRef]
- Mezzasoma, L.; Talesa, V.N.; Romani, R.; Bellezza, I. ANP and BNP exert anti-inflammatory action via NPR-1/cGMP axis by interfering with canonical, non-canonical, and alternative routes of inflammasome activation in human THP1 cells. Int. J. Mol. Sci. 2021, 22, 24. [Google Scholar] [CrossRef]
- Lucas, K.A.; Pitari, G.M.; Kazerounian, S.; Ruiz-Stewart, I.; Park, J.; Schulz, S.; Chepenik, K.P.; Waldman, S.A. Guanylyl cyclases and signaling by cyclic GMP. Pharm. Rev. 2000, 52, 375–414. [Google Scholar]
- Derbyshire, E.R.; Marletta, M.A. Structure and regulation of soluble guanylate cyclase. Annu. Rev. Biochem. 2012, 81, 533–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potter, L.R. Guanylyl cyclase structure, function and regulation. Cell. Signal. 2011, 23, 1921–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-N.; Watson, G.; Zhao, L. Cyclic guanosine monophosphate signalling pathway in pulmonary arterial hypertension. Vasc. Pharm. 2013, 58, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Irving, H.R.; Cahill, D.M.; Gehring, C. Moonlighting proteins and their role in the control of signaling microenvironments, as exemplified by cGMP and phytosulfokine receptor 1 (PSKR1). Front. Plant Sci. 2018, 9, 415. [Google Scholar] [CrossRef]
- Yuan, P.; Jauregui, E.; Du, L.; Tanaka, K.; Poovaiah, B. Calcium signatures and signaling events orchestrate plant–microbe interactions. Curr. Opin. Plant Biol. 2017, 38, 173–183. [Google Scholar] [CrossRef]
- Turek, I.; Irving, H. Moonlighting proteins shine new light on molecular signaling niches. Int. J. Mol. Sci. 2021, 22, 1367. [Google Scholar] [CrossRef]
- Wong, A.; Gehring, C.; Irving, H.R. Conserved functional motifs and homology modeling to predict hidden moonlighting functional sites. Front. Bioeng. Biotechnol. 2015, 3, 82. [Google Scholar] [CrossRef] [Green Version]
- Mitkiewicz, M.; Kuropatwa, M.; Kurowska, E.; Gorczyca, W.A. Different effects of soluble and particulate guanylyl cyclases on expression of inflammatory cytokines in rat peripheral blood mononuclear cells. Immunobiology 2011, 216, 423–430. [Google Scholar] [CrossRef]
- Arias-Diaz, J.; Garcia-Verdugo, I.; Casals, C.; Sanchez-Rico, N.; Vara, E.; Balibrea, J.L. Effect of surfactant protein A (SP-A) on the production of cytokines by human pulmonary macrophages. Shock 2000, 14, 300–306. [Google Scholar] [CrossRef]
- Arias-Diaz, J.; Vara, E.; Garcia, C.; Villa, N.; Balibrea, J.L. Evidence for a Cyclic Guanosine Monophosphate—Dependent, Carbon Monoxide—Mediated, Signaling System in the Regulation of TNF-α Production by Human Pulmonary Macrophages. Arch. Surg. 1995, 130, 1287–1293. [Google Scholar] [CrossRef]
- Broderick, K.E.; Zhang, T.; Rangaswami, H.; Zeng, Y.; Zhao, X.; Boss, G.R.; Pilz, R.B. Guanosine 3′, 5′-cyclic monophosphate (cGMP)/cGMP-dependent protein kinase induce interleukin-6 transcription in osteoblasts. Mol. Endocrinol. 2007, 21, 1148–1162. [Google Scholar] [CrossRef] [Green Version]
- Kalra, D.; Baumgarten, G.; Dibbs, Z.; Seta, Y.; Sivasubramanian, N.; Mann, D.L. Nitric oxide provokes tumor necrosis factor-α expression in adult feline myocardium through a cGMP-dependent pathway. Circulation 2000, 102, 1302–1307. [Google Scholar] [CrossRef] [Green Version]
- Aizawa, T.; Wei, H.; Miano, J.M.; Abe, J.-I.; Berk, B.C.; Yan, C. Role of phosphodiesterase 3 in NO/cGMP-mediated antiinflammatory effects in vascular smooth muscle cells. Circ. Res. 2003, 93, 406–413. [Google Scholar] [CrossRef] [Green Version]
- Borán, M.S.; Baltrons, M.A.; García, A. The ANP-cGMP-protein kinase G pathway induces a phagocytic phenotype but decreases inflammatory gene expression in microglial cells. Glia 2008, 56, 394–411. [Google Scholar] [CrossRef]
- Connelly, L.; Jacobs, A.T.; Palacios-Callender, M.; Moncada, S.; Hobbs, A.J. Macrophage endothelial nitric-oxide synthase autoregulates cellular activation and pro-inflammatory protein expression. J. Biol. Chem. 2003, 278, 26480–26487. [Google Scholar] [CrossRef] [Green Version]
- Kiemer, A.K.; Hartung, T.; Vollmar, A.M. cGMP-mediated inhibition of TNF-α production by the atrial natriuretic peptide in murine macrophages. J. Immunol. 2000, 165, 175–181. [Google Scholar] [CrossRef] [Green Version]
- Ladetzki-Baehs, K.; Keller, M.; Kiemer, A.K.; Koch, E.; Zahler, S.; Wendel, A.; Vollmar, A.M. Atrial natriuretic peptide, a regulator of nuclear factor-κB activation in vivo. Endocrinology 2007, 148, 332–336. [Google Scholar] [CrossRef] [Green Version]
- Moriyama, N.; Taniguchi, M.; Miyano, K.; Miyoshi, M.; Watanabe, T. ANP inhibits LPS-induced stimulation of rat microglial cells by suppressing NF-κB and AP-1 activations. Biochem. Biophys. Res. Commun. 2006, 350, 322–328. [Google Scholar] [CrossRef] [Green Version]
- Siednienko, J.; Nowak, J.; Moynagh, P.N.; Gorczyca, W.A. Nitric oxide affects IL-6 expression in human peripheral blood mononuclear cells involving cGMP-dependent modulation of NF-κB activity. Cytokine 2011, 54, 282–288. [Google Scholar] [CrossRef]
- von Bülow, V.; Rink, L.; Haase, H. Zinc-mediated inhibition of cyclic nucleotide phosphodiesterase activity and expression suppresses TNF-α and IL-1β production in monocytes by elevation of guanosine 3′, 5′-cyclic monophosphate. J. Immunol. 2005, 175, 4697–4705. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Yan, L.; Wesley, R.A.; Danner, R.L. Nitric oxide increases tumor necrosis factor production in differentiated U937 cells by decreasing cyclic AMP. J. Biol. Chem. 1997, 272, 5959–5965. [Google Scholar] [CrossRef] [Green Version]
- Xiong, H.; Zhu, C.; Li, F.; Hegazi, R.; He, K.; Babyatsky, M.; Bauer, A.J.; Plevy, S.E. Inhibition of interleukin-12 p40 transcription and NF-κB activation by nitric oxide in murine macrophages and dendritic cells. J. Biol. Chem. 2004, 279, 10776–10783. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.H.; Choi, D.H.; Song, K.S.; Shin, K.H.; Chun, B.G. Zaprinast, an inhibitor of cGMP-selective phosphodiesterases, enhances the secretion of TNF-α and IL-1β and the expression of iNOS and MHC class II molecules in rat microglial cells. J. Neurosci. Res. 2002, 67, 411–421. [Google Scholar] [CrossRef]
- García-Ortiz, A.; Serrador, J.M. Nitric oxide signaling in T cell-mediated immunity. Trends Mol. Med. 2018, 24, 412–427. [Google Scholar] [CrossRef]
- Gnipp, S.; Mergia, E.; Puschkarow, M.; Bufe, A.; Koesling, D.; Peters, M. Nitric oxide dependent signaling via cyclic GMP in dendritic cells regulates migration and T-cell polarization. Sci. Rep. 2018, 8, 10969. [Google Scholar] [CrossRef] [Green Version]
- Domon, H.; Honda, T.; Oda, T.; Yoshie, H.; Yamazaki, K. Early and preferential induction of IL-1 receptor-associated kinase-M in THP-1 cells by LPS derived from Porphyromonas gingivalis. J. Leukoc. Biol. 2008, 83, 672–679. [Google Scholar] [CrossRef]
- Schlossmann, J.; Schinner, E. cGMP becomes a drug target. Naunyn-Schmiedebergs Arch. Pharmacol. 2012, 385, 243–252. [Google Scholar] [CrossRef] [Green Version]
- Murad, F. Nitric oxide and cyclic GMP in cell signaling and drug development. N. Engl. J. Med. 2006, 355, 2003–2011. [Google Scholar] [CrossRef]
- Peng, H.-B.; Libby, P.; Liao, J.K. Induction and stabilization of IκBα by nitric oxide mediates inhibition of NF-κB. J. Biol. Chem. 1995, 270, 14214–14219. [Google Scholar] [CrossRef] [Green Version]
- Vollmar, A.M. The role of atrial natriuretic peptide in the immune system. Peptides 2005, 26, 1086–1094. [Google Scholar] [CrossRef]
- Kim, M.-Y.; Park, J.-H.; Mo, J.-S.; Ann, E.-J.; Han, S.-O.; Baek, S.-H.; Kim, K.-J.; Im, S.-Y.; Park, J.-W.; Choi, E.-J. Downregulation by lipopolysaccharide of Notch signaling, via nitric oxide. J. Cell Sci. 2008, 121, 1466–1476. [Google Scholar] [CrossRef] [Green Version]
- Morita, R.; Uchiyama, T.; Hori, T. Nitric oxide inhibits IFN-α production of human plasmacytoid dendritic cells partly via a guanosine 3′, 5′-cyclic monophosphate-dependent pathway. J. Immunol. 2005, 175, 806–812. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Hou, Y.; Shen, J.; Mehra, R.; Kallianpur, A.; Culver, D.A.; Gack, M.U.; Farha, S.; Zein, J.; Comhair, S. A network medicine approach to investigation and population-based validation of disease manifestations and drug repurposing for COVID-19. PLoS Biol. 2020, 18, e3000970. [Google Scholar] [CrossRef]
- Flam, B.R.; Eichler, D.C.; Solomonson, L.P. Endothelial nitric oxide production is tightly coupled to the citrulline–NO cycle. Nitric Oxide 2007, 17, 115–121. [Google Scholar] [CrossRef]
- Pantazi, I.; Al-Qahtani, A.A.; Alhamlan, F.S.; Alothaid, H.; Matou-Nasri, S.; Sourvinos, G.; Vergadi, E.; Tsatsanis, C. SARS-CoV-2/ACE2 interaction suppresses IRAK-M expression and promotes pro-inflammatory cytokine production in macrophages. Front. Immunol. 2021, 12, 683800. [Google Scholar] [CrossRef]
- del Fresno, C.; Gómez-García, L.; Caveda, L.; Escoll, P.; Arnalich, F.; Zamora, R.; López-Collazo, E. Nitric oxide activates the expression of IRAK-M via the release of TNF-α in human monocytes. Nitric Oxide 2004, 10, 213–220. [Google Scholar] [CrossRef]
- Mao, K.; Chen, S.; Chen, M.; Ma, Y.; Wang, Y.; Huang, B.; He, Z.; Zeng, Y.; Hu, Y.; Sun, S. Nitric oxide suppresses NLRP3 inflammasome activation and protects against LPS-induced septic shock. Cell Res. 2013, 23, 201–212. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.-M.; Talanian, R.V.; Li, J.; Billiar, T.R. Nitric oxide prevents IL-1β and IFN-γ-inducing factor (IL-18) release from macrophages by inhibiting caspase-1 (IL-1β-converting enzyme). J. Immunol. 1998, 161, 4122–4128. [Google Scholar]
- VerPlank, J.J.; Tyrkalska, S.D.; Fleming, A.; Rubinsztein, D.C.; Goldberg, A.L. cGMP via PKG activates 26S proteasomes and enhances degradation of proteins, including ones that cause neurodegenerative diseases. Proc. Natl. Acad. Sci. USA 2020, 117, 14220–14230. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Geng, D.; Ciavattone, N.; Lasola, J.J.; Shrestha, R.; Sanchez, A.; Guo, J.; Vlk, A.; Younis, R.; Wang, L.; Brown, A.J.; et al. Induction of IRAKM in melanoma induces caspase-3 dependent apoptosis by reducing TRAF6 and calpastatin levels. Commun. Biol. 2020, 3, 306. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Zhang, Y.; Gu, L.; Lan, M.; Liu, C.; Wang, M.; Su, Y.; Ge, M.; Wang, T.; Yu, Y.; et al. Islr regulates canonical Wnt signaling-mediated skeletal muscle regeneration by stabilizing Dishevelled-2 and preventing autophagy. Nat. Commun. 2018, 9, 5129. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.H.; Axell, A.; Turek, I.; Wright, B.; Meehan-Andrews, T.; Irving, H.R. Modulation of Inflammatory Cytokine Production in Human Monocytes by cGMP and IRAK3. Int. J. Mol. Sci. 2022, 23, 2552. https://doi.org/10.3390/ijms23052552
Nguyen TH, Axell A, Turek I, Wright B, Meehan-Andrews T, Irving HR. Modulation of Inflammatory Cytokine Production in Human Monocytes by cGMP and IRAK3. International Journal of Molecular Sciences. 2022; 23(5):2552. https://doi.org/10.3390/ijms23052552
Chicago/Turabian StyleNguyen, Trang H., Anna Axell, Ilona Turek, Bree Wright, Terri Meehan-Andrews, and Helen R. Irving. 2022. "Modulation of Inflammatory Cytokine Production in Human Monocytes by cGMP and IRAK3" International Journal of Molecular Sciences 23, no. 5: 2552. https://doi.org/10.3390/ijms23052552